임상시험용의약품 (Investigational medicinal products)의 품질 자료를 준비하다가 보면 꼭 접하게 되는 IMPD와 CTD이다. 이 두 가지 품질 자료 종류는 형식도 비슷해보이는데 도대체 뭐가 다른 것인지 알아보고 IMPD의 구성에 대해 더 자세히 알아보겠다.
정의
IMPD : Investigational Medicinal Product Dossier 의 약자이다. 임상시험용의약품 품질문서를 뜻하며 유럽(EU)의 임상시험계획승인신청(CTA) 단계에서 제출하는 품질자료를 주로 IMPD로 지칭한다. IMPD는 일반적으로는 품질 자료 위주로 구성되어 있고 임상시험승인신청 단계에서 제출되는 자료이다.
CTD : Common Technical Document의 약자이다. 국제공통기술문서라는 뜻으로, 허가 신청 시 국제적으로 통일된 자료 제출을 위해 만들어진 형식이다. 2000년대 초에 미국, 유럽, 일본 등의 허가 제출 시 적용되고 있으며 한국은 신약부터 순차적으로 CTD 작성 의무화가 적용되고 있다. CTD에는 품질 뿐만 아니라 개요, 비임상, 임상 자료 등을 포함하는 총 5개의 Module로 구성되어 있다. 미국에는 임상신청(IND) 단계에서도 CTD 형식으로 제출된다.
*한국에서는 임상시험계획승인(IND) 신청 시 '임상시험용의약품 품질문서 작성 가이드라인'에 따라 품질자료를 제출하면 되고, 위의 IMPD, CTD 형식도 해당 내용을 포함하고 있으므로 제출이 가능하다.
2023.02.17 - [직장생활/의약품 RA 업무 실습] - 의약품 임상시험계획승인(IND) [1] 제출자료 범위
IMPD 형식
IMPD는 원료(Drug Substance), 완제(Drug Product), 부록(Appendix)의 3가지 파트로 크게 구성되어 있다.
각 섹션과 명칭을 알아보겠다.
2.2.1.S Drug Substance
2.2.1.S.1 General information
2.2.1.S.1.1 Nomenclature
2.2.1.S.1.2 Structure
2.2.1.S.1.3 General properties
2.2.1.S.2 Manufacture
2.2.1.S.2.1 Manufacturer(s)
2.2.1.S.2.2 Description of manufacturing process and process controls
2.2.1.S.2.3 Control of materials
2.2.1.S.2.4 Control of critical steps and intermediates
2.2.1.S.2.5 Process validation and/or evaluation
2.2.1.S.2.6 Manufacturing process development
2.2.1.S.3 Characterisation
2.2.1.S.3.1 Elucidation of structure and other characteristics
2.2.1.S.3.2 Impurities
2.2.1.S.4 Control of the Drug Substance
2.2.1.S.4.1 Specification(s)
2.2.1.S.4.2 Analytical procedures
2.2.1.S.4.3 Validation of analytical procedures
2.2.1.S.4.4 Batch analyses
2.2.1.S.4.5 Justification of specification(s)
2.2.1.S.5 Reference standards or materials
2.2.1.S.6 Container closure system
2.2.1.S.7 Stability
2.2.1.P Investigational medicinal product under test
2.2.1.P.1 Description and composition of the investigational medicinal product
2.2.1.P.2 Pharmaceutical development
2.2.1.P.2.1 Manufacturing process development
2.2.1.P.3 Manufacture
2.2.1.P.3.1 Manufacturer(s)
2.2.1.P.3.2 Batch formula
2.2.1.P.3.3 Description of manufacturing process and process controls
2.2.1.P.3.4 Controls of critical steps and intermediates
2.2.1.P.3.5 Process validation and/or evaluation
2.2.1.P.4 Control of excipients
2.2.1.P.4.1 Specifiacations
2.2.1.P.4.2 Analytical procedures
2.2.1.P.4.3 Validation of the analytical procedures
2.2.1.P.4.4 Justification of specifications
2.2.1.P.4.5 Excipients of animal or human origin
2.2.1.P.4.6 Novel excipients
2.2.1.P.5 Control of the investigational medicinal product
2.2.1.P.5.1 Specifications
2.2.1.P.5.2 Analytical procedures
2.2.1.P.5.3 Validation of analytical procedures
2.2.1.P.5.4 Batch analyses
2.2.1.P.5.5 Characterisation of impurities
2.2.1.P.5.6 Justification of specification(s)
2.2.1.P.6 Reference standards or materials
2.2.1.P.7 Container closure system
2.2.1.P.8 Stability
7. Appendices
7.2.1.A.1 Facilities and equipment
7.2.1.A.2 Adventitious agents safety evaluation
7.2.1.A.3 Novel excipients
7.2.1.A.4 Solvents for reconstitution and diluents
시험약은 위의 DS(S) 및 DP(P) 파트로 구성된다.
만약 임상시험계획서 디자인에 위약(Placebo)이 있는 경우 위약에 대한 P 파트가 추가로 제출되어야 한다.
대조약(comparator products)을 사용하는 경우에는 해당 대조약이 허가된 의약품이라면 해당 SmPC (Summary of Product Characteristics) 문서를 제출하면 되지만, 눈가림을 유지하기 위해 시험약과 성상이 유사하게끔 추가 제조공정이나 포장공정이 수행된 경우에는 modified 된 정도에 따라 simplified IMPD를 제출하거나 대조약에 대한 IMPD P 파트를 추가로 제출하여야 한다.
또한 임상시험용의약품을 개발하다가 품질에 변경사항이 있을 수 있는데 이 때 변경신청(Substantial modification)이 필요한지와 그에 대한 요구사항은 아래와 같다. 출처의 가이드라인 맨 하단의 Table 1 - 3에서도 확인할 수 있다.
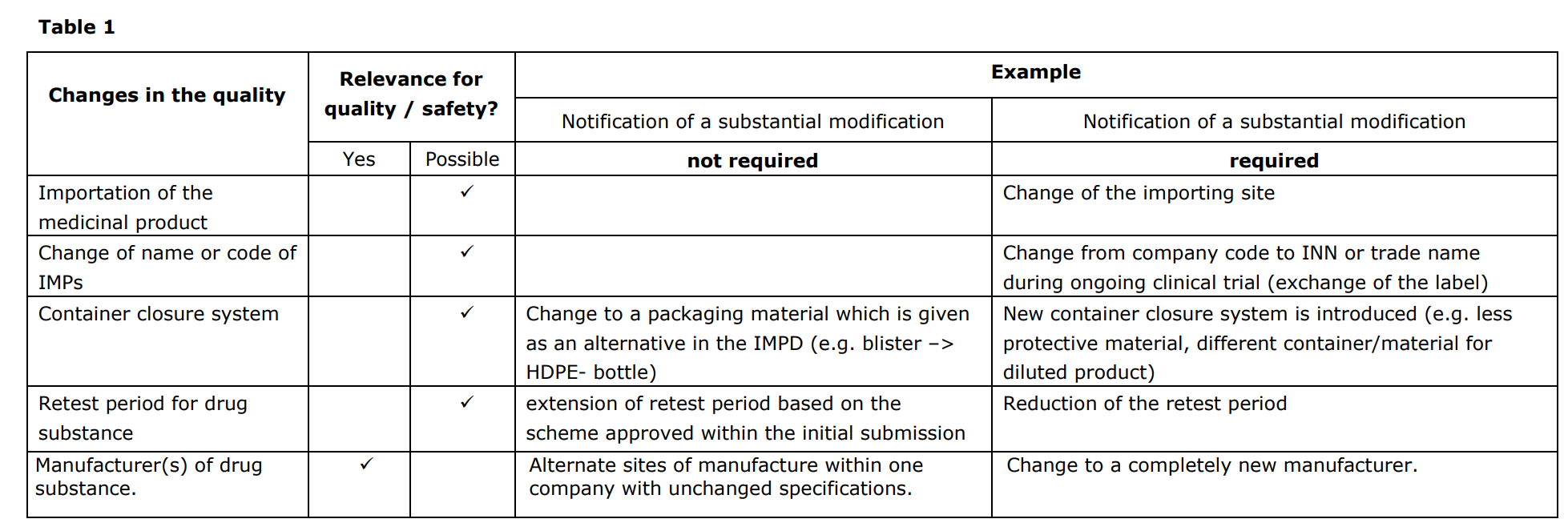
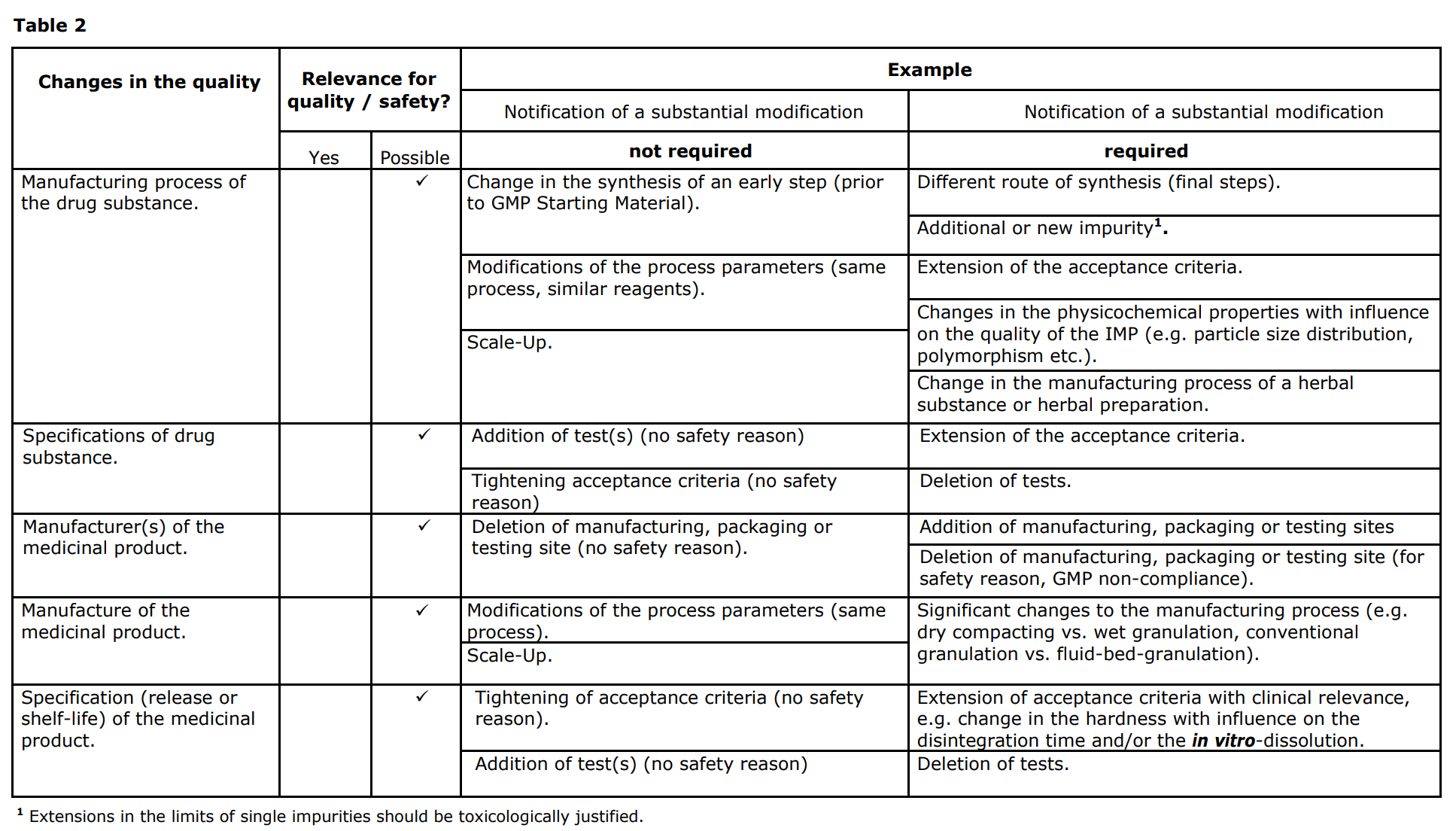
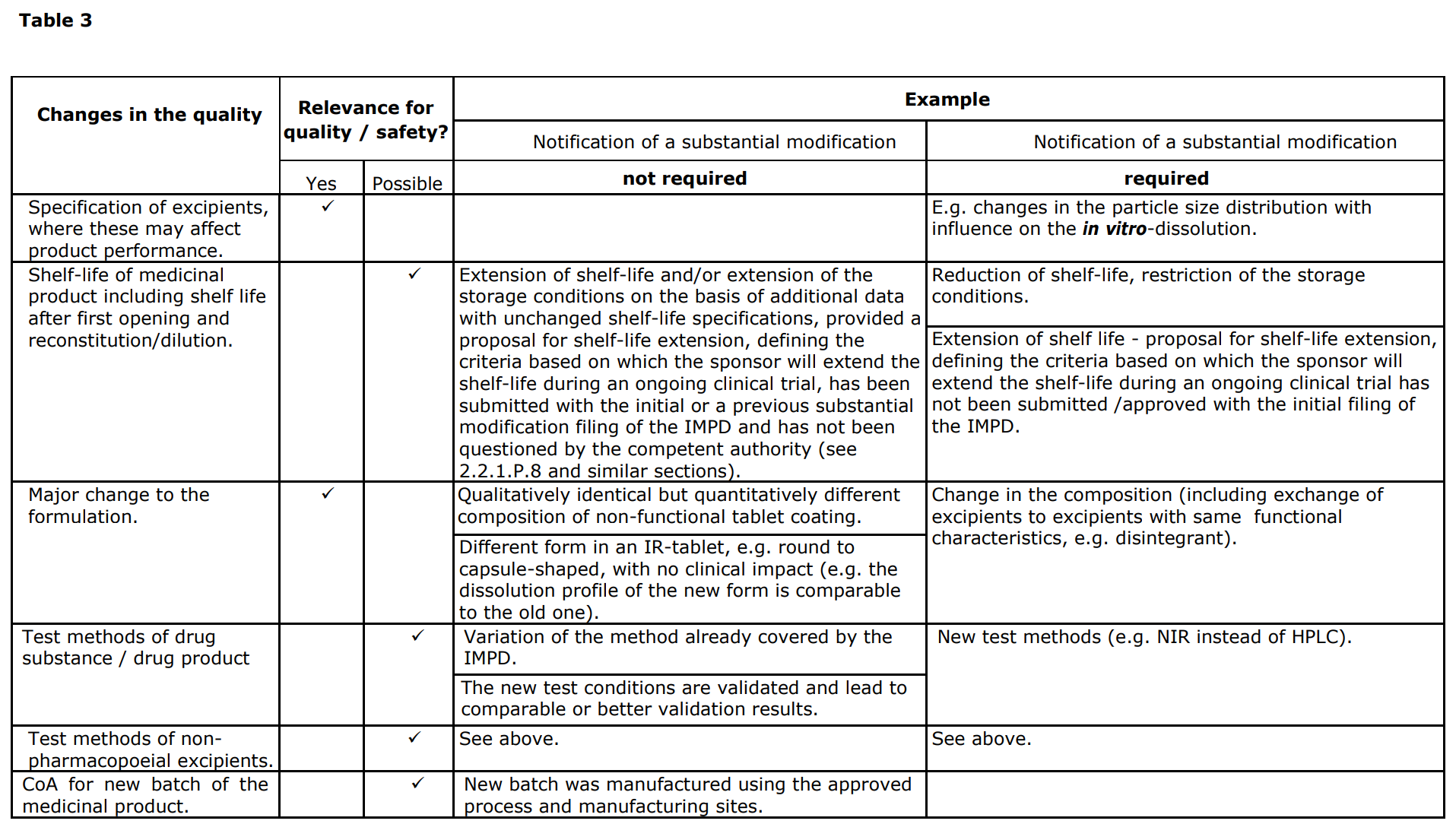
출처: EMA guideline : Guideline on the requirements for the chemical and pharmaceutical quality documentation concerning investigational medicinal products in clinical trials (20 September 2017, EMA/CHAMP/QWP/545525/2017)
'의약품 인허가' 카테고리의 다른 글
| 2024년도 새해 맞이 임상시험 실시상황 보고는 필수인거 알죠? (4) | 2024.01.04 |
|---|---|
| 반추동물 유래성분의 TSE 미감염 관련 제출자료 (0) | 2023.04.10 |
| [신약] 보술리프정(보수티닙일수화물) 허가 보고서 살펴보기 (0) | 2023.04.04 |
| 의약품 임상시험계획승인(IND) 절차 (0) | 2023.03.31 |
| 의약품 품목허가 제출서류 국문(영문) 정리 (0) | 2023.03.30 |




댓글