지난 포스팅에 이어서 의약품안전나라 홈페이지에서 IND 신청서를 작성하는 방법을 알아보자. 편의를 위해 국내 A회사에서 A-1234라는 임상시험용의약품(신약, 캡슐제)에 대해 미국 FDA 에 1상 IND 승인을 받은 후 국내에도 1상 임상시험계획승인 신청을 한다라는 가상의 설정으로 설명하겠다.
1. 신청내역

- 원개발사: 현재 임상시험용의약품 중 시험약의 개발권을 가지고 있는 회사를 말하며, 신청인과 같을수도, 다를수도 있다. 같다고 가정하에 A회사 명칭, 국가, 소재지를 입력해준다.
- 제품명, 성분명: 임상단계에서 제품명과 성분명은 A-1234와 같이 코드명으로 되어 있는 경우가 많고, 이후 품목허가 단계에서는 실제 유통될 제품명과 상세 성분명으로 신청하여야 한다. 지금은 임상단계이니 코드명으로 입력하였다.
- 임상시험계획승인을 위한 제출자료의 범위: IND 포스팅 [1]탄에서 살펴보았던 제출자료 범위에 따라 선택해준다. A-1234가 이전과 다른 전혀 새로운 성분의 신약이라는 가정을 하고 개발중인 신약으로 선택한다.
- 국내허가여부: 아직 1상 임상시험 신청 단계이므로 국내 허가여부는 아니오로 선택한다.
- 마약류분류: 마약류로 분류되는 성분이라면 마약/항정/한외마약/대마/원료물질 중 선택을 한다. A-1234는 마약류가 아닌 것으로 가정하에 선택하지 않았다.
- 타국가 시판허가 여부: 1상 임상시험 단계이므로 아니오를 선택하였다.
- 타국가 임상시험계획 승인 여부: 미국 FDA에 1상 IND 승인 완료 가정하여 예로 선택하였고 승인 국가명을 미국으로 선택하였다.
- 의약품 형태: 합성원료로 만든 캡슐제로 가정하였으므로 합성의약품으로 선택하였다.
- 사용(유효)기간: 제조일로부터 OO개월 형식으로 입력한다. 이 부분은 CMC 자료의 안정성시험 결과를 보고 입력해야 하는데, 24개월까지 모든 안정성시험 결과가 나왔다면 "제조일로부터 24개월"로 입력하고, 24개월까지로 계획하였으나 아직 완료되지 않은 상태라면 자료요건 확인 후 "제조일로부터 최대 24개월 (안정성시험계획에 따라 자체 검사)"로 입력해야 한다. 이번에는 24개월 자료가 있다는 가정 하에 작성했다.
- 자료보호여부: 이건 자세히는 모르겠으나 기밀자료로 "예"라고 선택했다.
- 수탁기관 정보: 임상시험 진행에 필요한 수탁기관이 있다면 정보를 기재한다.
2. 제조원

A회사에서 자체적인 GMP 시설을 가지고 있어서 자사제조 형태로 모든 공정을 진행한다는 가정하에, 제조소 버튼을 선택 후 제조원을 조회하여 입력한다. 소재지 뒤에는 수행공정을 괄호로 입력한다. 예를들어, A-1234와 그 위약의 생산을 모두 진행한다면 (A-1234 및 위약 제조 및 포장) 등과 같이 입력한다. 여기서 주의할 점은, 제조원의 GMP 증명서 등 근거자료에 있는 명칭과 소재지 문구가 일치하도록 입력하여야 한다.
3. 원료/성분
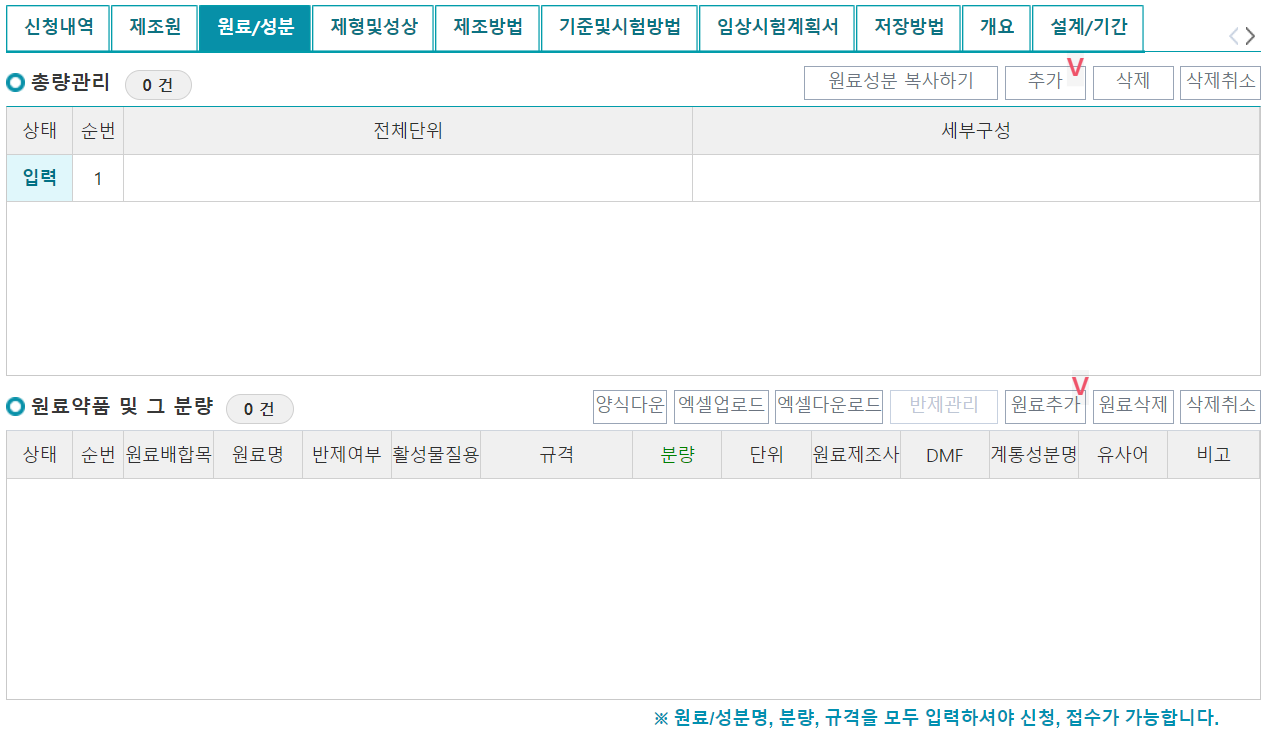
원료/성분 탭의 총량관리에서 "추가"버튼을 누르고 하나의 단위 당 각 원료약품 및 그 분량을 입력할 수 있다. 원료추가 버튼을 클릭하면 아래와 같이 팝업창이 뜨는데, A-1234의 주성분도 A-1234 코드를 사용한다는 가정하에 입력해보았다.

원료명과 배합목적을 선택하고, 반제품 형태가 아닌 것으로 가정하여 "아니오"를 선택했다. 개발중인 신약의 경우에는 대부분의 주성분이 별규 규격을 따르는데, 이 경우 별규로 규격선택 후 별규문서를 첨부해야 한다. 분량과 단위를 입력하면 끝. 활성물질, 원료제조사명, DMF는 해당 시 작성한다. 또한, 위와 같이 주성분을 추가했다면 각 첨가제를 하나씩 동일한 방법으로 추가해준다.
4. 제형 및 성상

제형 탭에서 "추가" 버튼을 누른 후 제형명을 "조회"하여 해당되는 제형을 클릭하면 자동으로 코드와 함께 입력된다. 성상 탭에서 "추가" 버튼을 누른 후 아래 박스에 내용을 입력하면 자동으로 위의 입력 칸에 저장된다.
5. 파일첨부 (제조방법, 기준 및 시험방법, 임상시험계획서)

제조방법, 기준 및 시험방법, 임상시험계획서는 구비한 파일을 "파일첨부" 버튼을 클릭하여 첨부한다.
6. 저장방법

저장방법 탭에서는 안정성시험결과를 근거로 보관조건을 기재한다.
7. 개요

개요 탭부터는 제출하는 임상시험계획서를 보고 정확한 정보를 입력하면 된다. 위에 생략한 칸의 경우 임상시험계획서에 작성된 내용은 모두 기입한다. 각 항목을 클릭해보면 용어 검색도 가능하다.
- 임상시험제목: 실시하고자 하는 임상시험의 목적 및 내용을 포함한 임상시험 계획서의 공식적인 제목
- 임상시험의 목적: 임상시험을 수행하는 주된 목적으로, 중재나 치료의 안전성, 유효성 등을 평가하는 것 등이 목적이 될 수 있음. 의약품의 경우 해당 의약품의 약동학·약력학적 특성, 안전성·내약성, 유효성 평가 등이 임상시험의 목적이 될 수 있음.
- 임상시험의 용도: 허가용 임상시험이란 의약품 품목 허가를 목적으로 하는 임상시험, 연구용 임상시험이란 의약품 품목 허가 취득 목적이 아닌 학문적 및 과학적 가설 검증을 위한 임상시험
- 국내/국외: 국내개발이란 개발 주체가 국내 제약사 등에서 개발한 의약품을 말하고, 국외개발이란 개발 주체가 국외 제약사 등에서 개발한 의약품을 말함
- 상세구분: 다국가/국내/가교시험 중 선택한다.
- 다국가: 제약사 등이 의약품을 개발하여 우리나라를 포함하여 2개국 이상에서 실시하는 임상시험
- 국내 임상시험: 제약사 등이 의약품을 개발하여 국내에서 실시하는 임상시험
- 가교시험: 국내 임상시험 중 의약품의 안전성·유효성에 관한 민족적 요인에 차이가 있어 외국 임상자료를 그대로 적용하기가 어려운 경우 국내에서 한국인을 대상으로 가교자료를 얻기 위하여 실시하는 시험 - 대상질환: 임상시험용의약품을 적용하여 안전성과 유효성 등을 확인할 수 있는 질환
- 대상질환명(적응증): 임상시험계획서 상의 대상질환명 또는 목표 적응증
8. 설계/기간
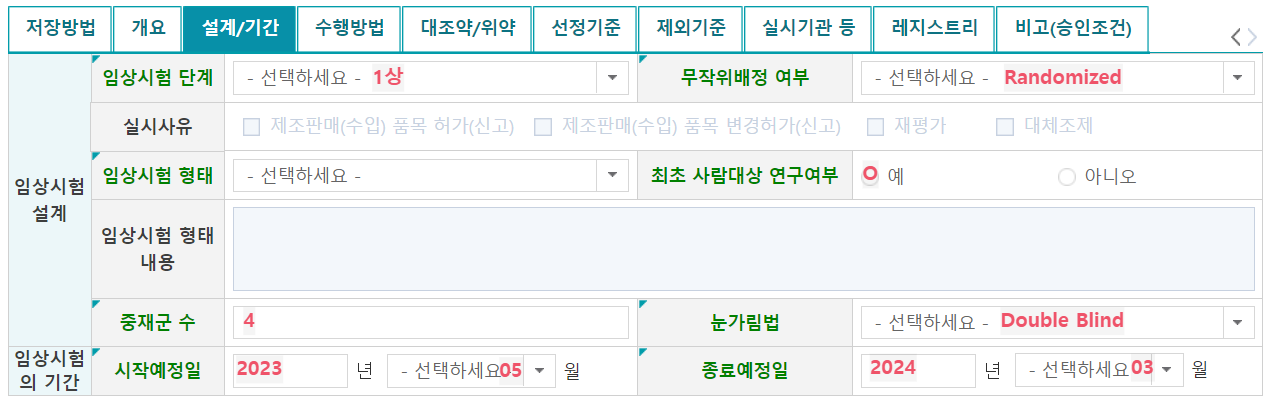
- 임상시험 단계: 0상, 1상, 2상, 3상 등 해당되는 단계를 선택
- 무작위배정 여부: 임상연구에 대상자를 배정하는 방식으로 할당 유형은 Randomized와 Non-randomized 2가지
- Randomized (무작위 배정): 임상시험 과정에서 발생할 수 있는 비뚤림(bias)을 줄이기 위해 연구자의 중재 없이 확률의 원리에 따라 대상자를 각 시험군 또는 대조군에 배정하는 과정
- Non-randomized (비 무작위 배정): 대상자를 치료군 또는 의약품 군에 배정할 때 무작위배정을 실시하지 않고, 어떤 기준에 따라 배정하도록 설계하는 방법 - 임상시험 형태: 중재군을 배치하는 방법
- Single (단일): 모든 대상자가 동일한 하나의 중재를 받는 설계 방식
- Parallel (병행): 대상자가 2개 이상의 군 중 한 군에 배정되어 치료를 받는 것. 이때 치료는 단일 용량 혹은 다용량의 시험약과 대조 치료를 말하며, 대조 치료에는 위약 혹은 치료효과가 있는 비교약물이나 표준약물이 포함됨
- Crossover (교차): 대상자가 2가지 이상의 치료에 순차적으로 배정되어 대상자가 치료군과 대조군 역할을 모두 하는 것
- Factorial (요인): 치료의 조합을 다양하게 하여 두 개 이상의 치료를 동시에 평가하는 방식 - 최초 사람대상 연구여부: 시험약이 인간에게 투여되는 최초의 제1상 임상시험
- 중재군 수: 연구를 위해 연구대상자에게 시험목적 및 설계에 따라 물리적 개입이 포함된 행위가 포함된 군
- 눈가림법: 임상시험에 관여하는 사람 또는 부서 등이 배정된 치료법/의약품에 대해 알지 못하도록 하는 절차
- Open (공개): 연구자와 대상자가 중재 배정에 대해 알고 있는 상태
- Single Blind (단일 눈가림): 연구 진행 과정에서 연구자와 대상자 중 한 쪽을 눈가림 상태로 유지하는 것. 일반적으로 대상자가 눈가림 유지 대상이 됨
- Double Blind (이중 눈가림): 대상자, 시험자, 모니터, 필요한 경우 자료 분석에 관여하는 자 등을 눈가림 상태로 하는 것
- Partial Blind (부분 눈가림): 부분 눈가림을 말함. 예) 관찰자 눈가림
임상시험 기간: 최초 스크리닝 시작 예정일부터 시험대상자 관련 자료 수집을 종료하는 예정일까지의 기간
9. 수행방법
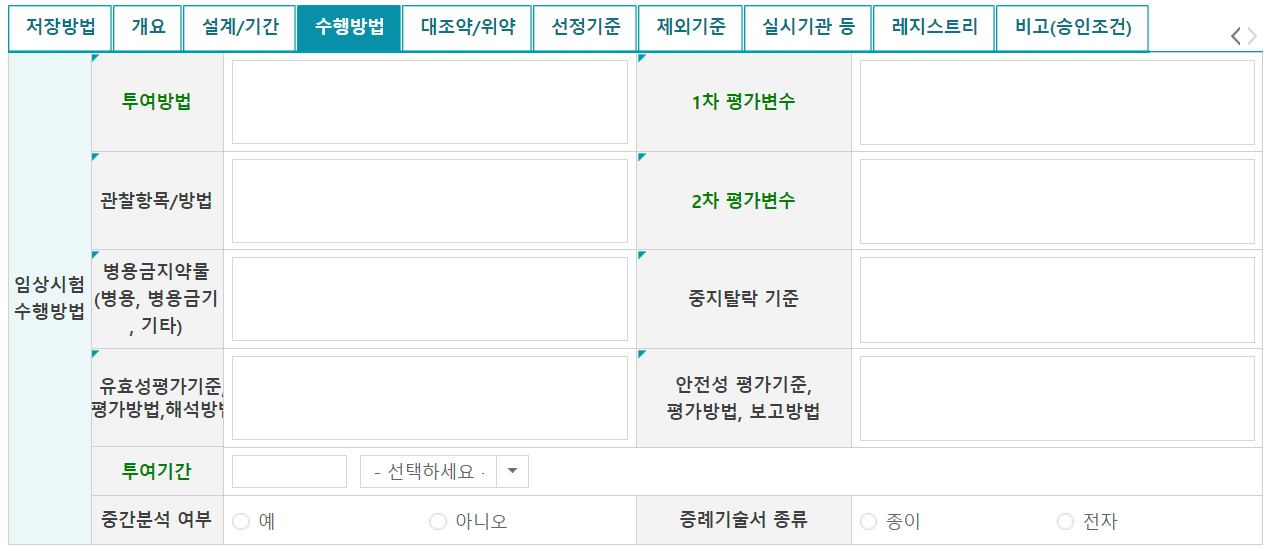
수행방법 탭의 경우에도 임상시험계획서를 근거로 작성한다.
- 투여기간: 임상시험계획서에 정의된 대상자별 최대 투여기간(F/U기간 포함)
- 중간분석 여부: 임상시험계획서 디자인 참고하여 선택
- 증례기술서 종류: CRF를 종이로 쓸건지, 전자로 쓸건지 선택
10. 대조약/위약

대조약 또는 위약이 있는 경우 체크 후 정보를 기재한다.
- 대조약: 시험약과 비교할 목적으로 사용되는 위약 또는 개발 중이거나 판매 중인 의약품
- 위약: 시험약의 효과를 확인하기 위해서 비교용으로 투여하는 의약품으로, 활성성분이 들어 있지 않아 효과를 나타내지 않는 약
11. 선정기준

건강한 성인을 대상으로하는 임상시험이라 가정할 때 시험대상자 선정기준은 모든 항목에 체크가 되어야 하고, 각 선정기준은 추가 탭을 통해 하나의 항목씩 입력한다. 선정기준이 방대하고 항목이 많은 경우에는 관련 항목끼리 묶어서 추가하기도 한다.
- 선정기준: 임상시험에 참여여부를 판단하는 자격기준 중 하나로서 개인이 임상시험에 참여하는데 필요한 기준 또는 그 목록
12. 제외기준

- 제외기준: 임상시험에 참여여부를 판단하는 자격기준 중 하나로서 개인을 임상시험으로부터 배제할 수 있는 기준 또는 그 목록
13. 실시기관 등

임상시험실시기관에 대해 각각 추가하여 작성한다. 1개의 기관이 될 수도 있고, 여러개의 기관이 될 수 있다. 임상시험 검체분석기관이 정해진 경우, 기관유형에서 선택하여 분석기관도 작성한다.
14. 레지스트리

국내 IND를 신청하고자하는 임상시험계획서가 해외에 먼저 승인된 경우 등 레지스트리가 등록된 경우가 있는데, 이 경우 레지스트리 명칭과 해당 임상시험의 등록번호를 기재한다. 레지스트리는 ClinicalTrials.gov (http://clinicaltrials.gov) 등에서 조회 가능하다. 다음 탭인 비고(승인조건)의 경우 잘 입력하지 않는 사항이라 생략하겠다.
15. 체크리스트 작성 및 구비서류 첨부
위의 신청서 입력과 더불어 IND 제출을 위해서는 구비서류를 첨부하여야 한다. 이 부분은 다음 포스팅에서 다뤄보도록 하겠다.
4탄 보기
'의약품 인허가' 카테고리의 다른 글
| 의약품 임상시험계획승인(IND) [5] 구비서류 업로드 방법 (0) | 2023.02.23 |
|---|---|
| 의약품 임상시험계획승인(IND) [4] 체크리스트 작성방법 (0) | 2023.02.22 |
| 의약품 임상시험계획승인(IND) [2] 의약품안전나라 로그인 후 민원신청까지 (0) | 2023.02.20 |
| 의약품 임상시험계획승인(IND) [1] 제출자료 범위 (0) | 2023.02.17 |
| 의약품 인허가(RA) 담당자의 북마크바 리스트 (0) | 2023.02.16 |




댓글